The following questions below address the Phil. Trans. R. Soc. B. paper entitled “The Evolution of HIV-1 and the Origin of AIDS” by Sharp and Hahn, 2010. These questions encouraged us to look further into the origin of the HIV virus and how it has evolved over time.
- What is phylogeography? How were the authors able to use phylogenetics and phylogeography to localize the origin of HIV strains?Phylogeography is the study of the historical processes that may be responsible for the contemporary geographic distribution of individuals. This is accomplished by considering the geographic distribution of individuals in light of the patterns associated with a gene genealogy. The authors were able to localize the origin of HIV strains by relating the virus to SIVs, which is the virus from non-human primates. Through phylogenetic analyses, the HIV strains were divided up into various groups, which were mixed among the SIVsmm lineages. Through this process they traced the source of HIV strains to a virus infecting the central subspecies of chimpanzees in a remote area in the southeast corner of Cameroon.
- How do they know that most HIV transmissions are intraspecific? Explain.
Most HIV transmissions are confined to members of the same species. This is based on the ways it can spread. HIV can only be passed through blood, bodily secretions or breast milk of an HIV-infected person therefore usually, but not limiting the spread of HIV, to within species. (source: CDC--HIV Transmission)
- What do the env, vpu, and nef genes code for in HIV? Would you then expect high levels of mutation rates to be tolerated in these genes? All of these genes code for accessory or structural proteins. Env, which stands for envelope, codes for a surface protein that enables the host cell to attach itself to the CD4 receptors present on the lymphocytes. Vpu is a integral membrane phosoprotein, which has many functions in the virus. It is responsible for CD4 degradation, as well as helping to release virions from infected cells. The Nef gene codes for a membrane associated phosoprotein. It also plays a role in the replication cycle of the virus and plays a role in cell apoptosis and in the virus’ ability to infect. One would expect very high levels of mutation in these genes because they deal with mostly replication and infection of new target cells. This is the main problem that researchers are dealing with when battling HIV. The virus is always finding novel ways to infect, replicate and find new target cells, while staying one step ahead of the researchers and their treatments.
- Apply Darwin’s postulates to the adaptation of the recombinant virus to the human host population.
 |
| The origin of the HIV virus |
Darwins postulates applied to HIV adaptation:
Variation: There are at least 12 known transmissions of SIV’s to humans, with hundreds to thousands of years of opportunities for this virus to have jumped back and forth between the chimpanzee-monkey- human hosts and vice versa. This has resulted in a large amount of variation within the virus itself: HIV-1 group M (most common form) has anti-CD4 activity and anti-tetherin activity, group N has only anti-tetherin activity, and group O has only anti-CD4 activity.
Variation is heritable: In order for these variations to spread and get passed onto offspring they must be heritable. These variations can be traced back to the ancestral form, SIVcpz virus and its inability to act against human tetherin. The transmission of SIV to human hosts occurred on three separate occasions from different ape species, resulting in various heritable adaptations amongst the virus groups to infect human hosts. The anti-CD4 activity was conserved from the ancestral SIV virus.
More offspring produced than able to survive: Many viral forms jumped from populations from ape to human host over the last thousand years, and only those well adapted to infect humans have remained. The HIV-1 group M, which accounts for perhaps 98% of HIV infections, has fully successful mutations to combat tetherin and CD4 activities resulting in the high rate of fitness of the virus.
Adaptation leads to evolution of the population: The transition of SIV to HIV resulted in selection pressures on the viral populations once transmitted from ape-human host. This pressure resulted in some strands of the virus to mutate in order to effect the human hosts more efficiently. This lead to those strands becoming more dominant in the population and to the evolution of the virus.
- What evidence does this study provide that we share an immediate common ancestor with Pan troglodytes?
Chimpanzees, Pan troglodytes, and humans are genetically very similar, as they are the two closest related species in the ape family. Chimpanzees were most likely infected by the SIV virus known to infect the red-capped mangabeys monkey population, which is also native to the west central Africa region where HIV-1’s ancestral form is thought to have originated. Similarly to the mutations brought about by the selection pressures in the transition from ape-humans, the mutations from the ancestral monkey form of SIVs to the current ape/chimpanzee form SIVgor are thought to have been due to the recombination of two separate viral strains of SIV. HIV-1 group M, the main form of AIDS infecting humans, has been traced back to SIV in chimpanzees, Pan troglodytes, in addition to other forms such as HIV-1 groups N, O, and P.
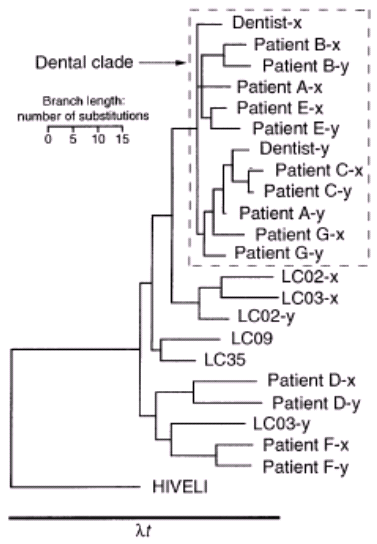
- Refer to Figure 1 which has been copied from the paper from Science entitled “Application and Accuracy of Molecular Phylogenies” (Hillis et al. 1994; Vol. 264: 671-677). In the study referenced, the authors considered the allegations of 7 patients (A-G) that they had contracted HIV from their dentist. Were their allegations correct? Describe how the authors might have generated this tree.
 |
| Figure 1 |
Based on the diagram, shown in Figure 1, the allegations from the patients found within the dental clade are believed to be correct. Patients A, B, C, E, and G are all found within the dental clade and all had a similar strain of HIV to that of the dentist. These patients would have gotten the virus from their dentist because they all share a similar HIV sequence with the dentist. Though HIV has a high rate of mutation, somewhere between the frequency of 10-8 and 10-4, the genome can still be sequenced and tested for similarities with the original strain coming from the dentist (Mansky). The x’s and y’s found in Figure 1 represent different mutations within the HIV virus. Since the patient’s HIV sequence more closely resembles that of the dentist than the local controls, they must have contracted the same strain of HIV that originated from the dentist. However, patient’s D and F, not found within the dental clade, did not contract HIV from their dentist. The strains of the HIV virus found in patients D and F resemble the strains also found in the local controls, labeled by LC in Figure 1. Since the strains from the local controls and these patients are more closely related in the phylogenetic tree they would have received the virus from an alternate source than the dentist.
By looking at the different sequences of the genomes in each person who tested positive for the HIV virus, phylogeneticists are able to piece together the pathways the virus took and which people actually contracted the virus from the dentist. There are several methods of constructing a phylogenetic tree including Neighbor Joining Method, Parsimony, Genetic Distance Method. In the case of the dentist, the Neighbor Joining Method would compare two patients or compare the dentist to the patient’s HIV sequence and run an algorithm to determine if the two subjects had a small yield. A small yield between the two would allow the phylogeneticists to determine they were neighbors and they would be joined in the phylogenetic tree. This tree shows the relationships between the patients and allows the tree to be created based on the similarities in the HIV sequences (Li.) Parsimony involves creating a tree that has the simplest evolutionary explanation with the fewest conjectures or nodes in the tree (Li). The simplest tree created by this method replicates the evolutionary changes and mutations the virus underwent and shows how it was most likely transferred from one person to another. The Genetic Distance Method also uses an algorithm and weighs the differences in the HIV strain according to how closely the strains can be related (Li). Genetic analysis helps determine how similar or different the strains are. This was most likely used to see which strains were more similar to the dentist than to the local controls. It is to my understanding that the Parsimony method was used to construct this phylogenetic tree since it is the most commonly used tree and it most accurately displays the evolutionary pathway of the HIV virus.
References:
Li, Yan. "How to Build a Phylogenetic Tree." 489 Bio. N.p., n.d. Web. 5 Apr. 2014. <http://guava.physics.uiuc.edu/~nigel/courses/598BIO/498BIOonline-essays/hw2/files/hw2_li.pdf>.
Excellent job! Your answers were very complete and answered the questions effectively, except that #5 could have more fully addressed the question. Still, great work.
ReplyDelete29 out of 30.
-Dr. Walker